结合AI计算,澳大利亚科学院乔世璋&焦研团队给出CO₂电还原的C2 选择性新理解

第一作者:李昊博, 姜云玲
通讯作者:乔世璋,焦研
通讯单位:阿德莱德大学
DOI:10.1021/jacs.3c03022
全文速览
设计高选择性的催化剂,将 CO₂ 电还原成多碳(C2 )燃料是迫切和重要的。然而,目前人们对 C2 物种的选择性理论知之甚少。在这里,澳大利亚科学院院士乔世璋&焦研教授团队首次报告了一种将量子化学计算、人工智能(AI)集群和实验相结合的方法,用于开发 C2 产物选择性和氧化铜基催化剂组成之间关系的模型。他们认为,结合理论计算、人工智能集群和实验,可以切实地建立复杂反应的描述符和选择性之间的关系。这些发现将有助于研究人员设计将二氧化碳电还原成碳 C2 产物的方法。
本文亮点
- 证明适度氧化的铜表面更有利于 C-C 耦联反应。
- 通过第一性原理热力学计算确定了不同金属掺杂成分下这种氧化状态的临界电位条件。
- 利用基于掺杂元素物理性质的多维标度(MDS)结果建立了实验法拉第效率和临界电位之间的倒火山关系。
- 展示了电催化剂的设计,以通过早期和晚期过渡金属的共掺杂策略选择性地生成 C2 产物。
背景介绍
铜是催化二氧化碳电还原反应(CRR)生成多碳产物的唯一元素。最近,氧化铜基(CuOx)催化剂得到了越来越多的关注,因为它们可以通过成分和结构工程来调节目标产品的产量和选择性。考虑到决定单碳(C1)或多碳(C2 )产物选择性的 C-C 偶联反应是一个热催化步骤,不涉及质子和电子的转移,催化剂材料和表面结构是决定性的。基于目前的认识,决定 CuOx 催化剂 CRR 对 C2 产物选择性的因素是 Cu 活性位点的几何结构和氧化状态。实验证据表明,在 CRR 的还原电势条件下,氧化 Cu 基(OD-Cu)表面是被还原的,独特的方形 Cu 位点结构和纳米晶粒影响反应选择性。然而,不同的 Cu 氧化态(OSCu)值的 Cu 原子在 CuOx 上表现出不同类型的产物选择性。OSCu = 0.35 到 1 的活性位点促进 C-C 偶联步骤生成 C2 物种,如乙烯或乙醇,而 OSCu = 2 的活性位点生成 C1 种。尽管调整 Cu 基催化剂的活性位点结构和 OSCu 为实现高 C2 选择性提供了一种切实可行的解决方案,但目前尚缺乏系统的认识。
理论计算对 CRR 催化剂的设计和筛选具有重要意义,特别是当机器学习加速方法的应用增加了可能的催化剂活性位点结构的数量时。目前的研究重点是计算吸附能作为一个便行的理论模型,通过火山型曲线关系来对应催化活性。然而,对催化选择性的理论理解研究很少,特别是像 CRR 这样的复杂反应。其中一个原因是吸附能明显依赖于表面活性位点的组成和结构。但活性位点结构可以在特定的电位、pH 和溶剂条件下热力学转化为不同的结构,从而影响反应路径和选择性。例如,在 CRR 实验中,Cu 基催化剂的表面为 Cu 端;即吸附活性位点仍然由 Cu 原子组成,而杂原子通过 Cu 原子间接影响催化作用。这种效应与反应条件有关,因此导致活性位点结构的变化,这是设计催化剂的一个现实困难。
因此,有必要开发新的理论模型和描述符,以更好地对应实验的选择性。通过第一性原理热力学计算,可以将相对稳定的表面结构与“实际”反应条件联系起来。结合 AI 分析及实验结果发现,理论计算出的不同表面结构之间的临界过渡条件可作为选择性的描述符。因此,这种方法对设计催化剂是有用的,因为它结合了实验条件对活性位点结构的影响。
图文解析
1. 从四方 CuO(001)表面模拟 Cu 的氧化态(OSCu)

图 1. 具有不同表面结构的 Cu 氧化态(OSCu)的基准
2. OSCu 对吸附性能和反应性的影响
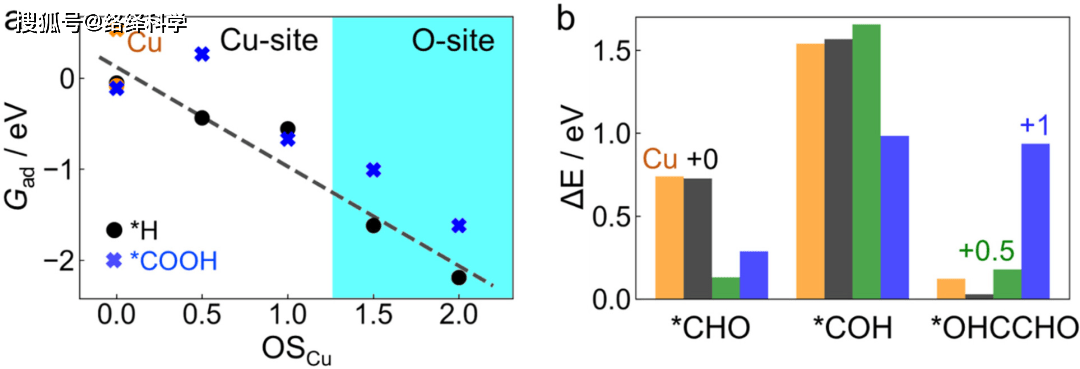
图 2. OSCu 调节的吸附性质和反应性
3. C–C 耦联机理

图 3. 电还原 CO₂ 生成 C2 产物的 C-C 偶联反应机理
4. 四方 CuO(001)表面的 Pourbaix 相图

图 4. 杂原子对四方 CuO(001)表面 Pourbaix 相图的影响
5. AI 集群方法为掺杂元素分类

图 5. 基于物理特性的掺杂元素分组
6. 近期实验结果的分析理解

图 6. (氧化)Cu 基催化剂 C2 产物的实验法拉第效率
总结与展望
在氧化铜基催化剂上,CRR 对 C2 产品的选择性与表面铜活性位点的氧化状态相关。这一点通过关键反应中间体的吸附性能的变化得到了证实。低氧化态 Cu 位点对 *CHO 的吸附弱于对 *CO 的吸附,这使得 *CO 到 *CHO 的步骤是不利的。因此,C-C 耦联是通过 *CO 二聚发生的。弱的 *CHO 吸附和不利的 *CO-*CO 二聚作用共同限制了 C2 产品的选择性。随着 Cu 氧化态的升高,*CO 到 *CHO 的步骤变得更加有利,C-C 耦联的步骤变为 *CHO 二聚。然而,随着 Cu 氧化态的进一步上升,*CHO 二聚逐渐变得不利。因此,可以得出结论,存在一个对 C-C 耦联最有利的适度氧化态,约 0.5。过渡金属掺杂有望改变 Cu 的氧化状态并影响 C2 产物的选择性。杂原子对表面 Cu 位点的氧化态的影响在基于第一性原理热力学计算的 Pourbaix 相图中得到证明,其中氧化态从 0 到 0.5 的临界电位条件是 C2 选择性的可靠描述符。
基于多达 30 种掺杂过渡金属元素的物理特性数据集的AI集群分类方法可以可靠地确定金属属于哪些区域。划分结果导致发现 C2 产物实验法拉第效率与临界电位条件之间的倒火山型关系。这可实际应用于选择元素来掺杂 Cu 基催化剂以生成 C2 产物。与本工作相比,此前 AI 辅助的 CRR 理论研究主要集中在预测吸附能上,并通过比较含碳中间体和含氢中间体的吸附能来推导与 HER 竞争的选择性。本研究将 AI 分析的目标从吸附能改为临界电位条件,并应用 AI 方法分析对 C2 产物的选择性,从而扩大了 AI 辅助催化剂设计的研究对象范围,有望为未来的研究提供参考。
总之,合理地结合 DFT 计算、AI 集群分类和实验结果分析,可以用来确定复杂反应的选择性和描述符之间的关系。这些发现将对研究人员在设计二氧化碳电还原转化为多碳 C2 产物的过程中具有重大意义。
文献来源
H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao, S.-Z. Qiao, J. Am. Chem. Soc. 2023. C2 Selectivity for CO₂ Electroreduction on Oxidized Cu-Based Catalysts. DOI: 10.1021/jacs.3c03022
作者介绍

乔世璋,现任澳大利亚科学院院士,澳大利亚阿德莱德大学化工学院纳米技术首席教授,能源与催化材料中心(Centre for Materials in Energy and Catalysis)主任,主要从事新能源技术纳米材料领域的研究,包括电池、电催化、光催化等。作为通讯联系人,在 Nature、Nature Energy、Nature Communications、Journal of American Chemical Society、Angewandte Chemie-International Edition、Advanced Materials 等国际顶级期刊发表学术论文超过 520 篇,引用超过 115,200 次,h 指数为 171。
乔世璋教授已获得多项重要奖励与荣誉,包括 2023 年澳大利亚研究理事会工业桂冠学者(ARC Industry Laureate Fellow),2021 年南澳年度科学家奖、2017 年 ARC 桂冠学者(ARC Australian Laureate Fellow)、2016 年埃克森美孚奖、2013 年美国化学学会能源与燃料部新兴研究者奖以及澳大利亚研究理事会杰出研究者奖(DORA)。乔教授是国际化学工程师学会会士、澳大利亚皇家化学会会士、英国皇家化学会会士等。同时,他担任国际刊物英国皇家化学会杂志 EES Catalysis 的主编,也是科睿唯安(Clarivate Analytics)/ 汤姆森路透(Thomson Reuters)化学、材料科学和环境与生态三个领域的高被引科学家(近十年有 120 篇高被引论文)。

焦研,副教授,阿德莱德大学化学工程学院副院长,澳大利亚研究理事会未来研究员。曾被评为澳大利亚 40 位科研新星之一,获澳大利亚政策与科学研究所颁发的杰出青年科学家奖,南澳 2021 年度杰出女性入围奖。
焦研主要研究兴趣为计算电催化,即用包含密度泛函理论和分子动力学在内的多尺度模拟来研究不同催化剂表面发生的电催化反应,并以此为基础来设计更新的催化剂。这些催化剂主要应用于清洁能源转换领域,涉及到的反应有氧还原,析氢,二氧化碳还原,氮还原和电池材料。课题组正在探索如何将机器学习和能源催化材料的计算结合起来。
已发表 100 余篇文章,共获超两万次引用,h 因子为 61。自 2019 年以来连续四年被科睿唯安评为化学方向的高被引学者。
声明:本文仅供科研分享,助力科学传播,不做盈利使用,如有侵权,请联系后台删除。
本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。